23/03/2009
Roberta Monteiro
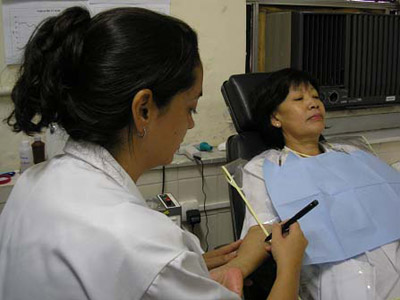
De forma pioneira no Brasil, um novo teste diagnóstico para fibrose cística, doença genética que compromete principalmente o trato digestivo e o aparelho respiratório, vem sendo implantado no Instituto Fernandes Figueira (IFF), unidade materno-infantil da Fiocruz. Trata-se da medida da diferença de potencial nasal (DPN), exame incluído nos critérios diagnósticos para a fibrose cística da Cystic Fibrosis Foundation (CFF), assim como o teste do suor, o teste do pezinho ampliado e a análise genética. O teste faz parte da segunda etapa do projeto Validação e reprodutibilidade da medida da diferença de potencial nasal, desenvolvido pelas médicas Izabela Sad e Laurinda Higa, do Serviço de Pneumologia do IFF.
|
|
| Com a participação dos voluntários que aceitarem realizar o exame será possível chegar à última etapa do projeto, que é a implantação formal do exame no IFF |
Segundo Izabela, o exame de DPN representa mais uma opção que possibilita o diagnóstico precoce da doença, alternativa que já vem sendo utilizada nos Estados Unidos, Canadá e em países da Europa, incluindo a Bélgica, local onde a pneumologista recebeu treinamento para a realização do teste e capacitação para sua implantação no país. “Como ainda não trabalhamos com o exame no Brasil, pretendemos a partir desse projeto criar um banco de dados com valores positivos e negativos, de forma a traçar o perfil da população em relação à doença. Isso será feito através dos resultados do exame de DPN feitos em três grupos compostos por pacientes com fibrose cística diagnosticada, pais de portadores da doença e pessoas saudáveis”, esclarece Izabela.
A médica explica que com a participação dos voluntários que aceitarem realizar o exame será possível chegar à última etapa do projeto, que é a implantação formal do exame no IFF - considerado o principal centro de referência (CR) para fibrose cística no Estado do Rio de Janeiro. A partir daí, será viável disponibilizar o exame de DPN aos pacientes do próprio Instituto, assim como a demanda externa do Rio de Janeiro, dos centros de referência dos demais estados do Brasil e dos países sulamericanos. Outra perspectiva é a ampliação do DPN para o resto do país por meio do treinamento de especialistas interessados na nova técnica.
O exame de DPN é um teste dinâmico, indolor, que dura cerca de 30 minutos, no qual se verifica, a nível celular, a função da proteína CFTR (proteína reguladora transmembrana da fibrose cística), cuja mutação genética é a principal responsável pelas características da doença. “Durante o exame, afere-se ao mesmo tempo o potencial elétrico no epitélio nasal e no antebraço do paciente, gerando assim uma diferença de potencial. No teste de DPN, o epitélio nasal é exposto a quatro tipos de soluções e de acordo com a reação a cada uma delas é possível obtermos o resultado do exame”, explica Izabela. Os interessados em participar do projeto como voluntários devem enviar um e-mail para izabela@iff.fiocruz.br.
Para a pneumologista do IFF Laurinda Higa, o novo teste de DPN deve ser indicado nos casos em que o teste do suor apresentar resultados normais e/ou duvidosos, associados a alguns sinais e sintomas sugestivos da doença. Vale ressaltar que mesmo com a implantação do exame de DPN, o teste do suor continua sendo o exame padrão-ouro para o diagnóstico da doença. Por ele dele é possível medir a quantidade de sódio e cloro no suor, já que uma das características dos portadores da fibrose cística é apresentar níveis altos de cloreto (sal) no suor. “Em pacientes com fibrose cística, quanto mais precocemente se estabelece o diagnóstico e tratamento, melhor o prognóstico”, afirma Laurinda.
A doença
A fibrose cística, também conhecida como mucoviscidose, é uma doença genética e não contagiosa, que causa alterações nas secreções de algumas glândulas do organismo. Estas secreções passam a ser produzidas com maior viscosidade e quantidade, comprometendo assim, o bom funcionamento de órgãos, como pulmões, fígado, pâncreas e intestino.
No trato respiratório, este muco espesso tende a se acumular nas vias aéreas, dificultando a passagem do ar e levando à retenção de bactérias. No trato digestivo, o aumento na viscosidade do muco pode levar à obstrução dos canais do pâncreas, dificultando a absorção de uma série de nutrientes pelo intestino. Este fato costuma provocar quadros clínicos caracterizados por diarreia crônica, desnutrição, má absorção dos nutrientes, gordura nas fezes e dificuldade de ganhar peso, além de sinusite de repetição, pneumonias crônicas por bactérias relacionadas à fibrose cística e disfunção pulmonar progressiva, podendo levar à insuficiência respiratória.
Segundo estimativa do Ministério da Saúde, aproximadamente 1,5 mil pessoas apresentam fibrose cística no Brasil. Isto equivale a uma criança acometida para cada 10 mil nascidas vivas. No IFF, encontram-se vinculados atualmente cerca de 130 crianças e adolescentes portadores da doença, que recebem tratamento e assistência multidisciplinar.
No futuro, quando a terapia gênica for realidade para esse grupo de pacientes, o DPN será indicado também como exame de monitoramento dessa terapêutica. Embora a doença ainda não tenha cura definitiva, o tratamento precoce em centro de referência, o acesso aos medicamentos e aos recursos laboratoriais, podem fazer toda a diferença no prognóstico e na qualidade de vida. Segundo o Ministério da Saúde, estes fatores contribuem muito para a redução da letalidade e o aumento da sobrevida dos pacientes, que vivem em média 40 anos nos países desenvolvidos e 18, nos países em desenvolvimento, como o Brasil.
O estudo faz parte do projeto Desenvolvimento e avaliação de tecnologias de diagnóstico da fibrose cística (FC) para o Sistema Único de Saúde (SUS), coordenado por Maria Virginia Peixoto e financiado pelos recursos da Rede de Pesquisa Clínica (PCL06) do Programa de Desenvolvimento Tecnológico em Saúde Pública-PDTSP da Vice-Presidência de Pesquisa e Desenvolvimento Tecnológico (VPPDT) da Fiocruz. A pesquisa recebe também recursos do projeto Desenvolvimento e avaliação de tecnologias de diagnóstico clínico e molecular da fibrose cística (FC), financiado pelo edital Pensa Rio da Faperj.
Publicado em 17/3/2009.